
The year 2021 has been a wild ride for structural biologists, with the advent of AlphaFold2, Deepmind’s solution to the 50-years old protein structure prediction problem, which won by a large margin the CASP14 competition in late 2020.
At the time, aside from its reported performance and the promise it held to solve protein structures challenging state-of-the-art experimental methods, nothing was said about how Deepmind would deliver its technology to the world. In summer 2021, Deepmind stated that it would release the source code of AlphaFold2 and actively collaborate with EMBL-EBI to produce what is now known as the AlphaFold Protein Structure Database (or AlphaFoldDB).
This database contains the prediction of AlphaFold2 for the human proteome, as well as 20 additional proteomes for other organisms. Since then, the license for the source code and the model parameters are available under the Apache 2.0 and Creative Commons Attribution 4.0 International (CC BY 4.0) licenses respectively.
Why is it important?
The protein structure prediction problem is a long-standing of structural biology. Proteins are mostly characterized by their sequence of amino acids, conventionally written from the amino-terminal extremity to the carboxyl-terminal extremity. However, proteins adopt different 3-dimensional shapes, which in turn characterize their functions.
It is also seen that protein of similar sequences tends to adopt the same overall shape, yet the opposite is not true: protein of similar shape can have vastly different sequences. It is expected that, as the sequence directly indicates the constituent of the proteins, then the overall protein structure is determined by this sequence.
While protein sequences can be obtained experimentally on a massive scale, it is much harder to resolve their 3D shape using empirical methods, which are highly impractical for industrial purposes such as drug development. Hence, several efforts have been made in order to predict protein structure in silico, using different methods such as homology modelling or ab initio predictions using physical models.
These approaches were not efficient enough to reach the level of precision of experimental methods. Recent approaches based on deep-learning, a machine-learning framework based on artificial neural network (ANN), showed promising improvements over the more historical methods.
It is then less of a surprise that Deepmind, a company who built its reputation on ANN algorithms surpassing human performance, came up with a solution of its own to this long-standing problem. AlphaFold2 and its competitors such as RoseTTAfold bring the promise of accelerated drug discovery and development by providing protein structure prediction with resolution almost as good as empirical methods.
This can be done by modelling the active molecules in drugs or pathogens. An often-cited example is the prediction of the structure of the protein ORF8, which is a protein expressed by SARS coronaviruses which is supposedly involved in immune invasion. In addition to structure prediction, AlphaFold2 has shown promising results for prediction protein complexes and can possibly predict protein-protein interactions in the broader sense.
Beyond the hype, there still remain improvements to be made for end-to-end in silico drug discovery: the overall current precision of AlphaFold on atomic positions prediction is not yet sufficient for accurate prediction of binding site. In addition to this, there are some other limitations: the model only output static models, and if many confirmations are outputed, it can be difficult to assess wether these actually exist in real biological systems.
Moreover, AlphaFold2 models are not reliable for intrinsically disordered proteins. Nevertheless, it is only a matter of time until these issues are solved.
AlphaFold2 in a nutshell
AlphaFold2 is an end-to-end solution to the protein structure prediction problem, starting from a sequence as an input, and protein structure model (PDB file format) as the output. Without going into the finer details, the sequence is first fed to a pre-processing pipeline whose only task is to obtain a multiple-sequence alignment (MSA) of all homologous sequences retrieved in different databases (such as Uniref90).
This MSA is then embedded in neural network architecture which output 3-dimensional models, which are then further relaxed using force fields calculation (AMBER). In addition to the models, AlphaFold2 outputs confidence scores to assess model quality.
Hurdles with running AlphaFold2
Using AlphaFold2 is rather straightforward, as it requires a minimal input file (in .fasta format), and few parameters. However, it is not possible to run it on a standard computer: the deep-learning model is optimized for GPU/TPU, and the pre-processing step with the MSA requires look-up in a collection of datasets up to 2.2 TB. Hence, only laboratories with High Performance Computing resources (HPC) can use AlphaFold2.
For research groups without such resources, it is also possible to use cloud-based services, such as Microsoft Azure, Google Cloud, or Amazon Web Services, albeit this solution can be costly if no precaution with resource allocation is taken. There exists solutions based on notebooks hosted on Google Colab, such as the official AlphaFold Colab notebook, or the ColabFold initiative, but these solutions have their shortcomings, as the resources freely provided are limited. Hence, it is not possible to run several jobs in batch.
At BioStrand, we opted for a solution based on AWS EC2 instances and EBS volumes for data storage. We have built an AlphaFold Docker image, so that the image can be stored then pulled from our Elastic Container Registry for direct use. We chose to store the 2.2 TB dataset on a volume of similar disk size, and we made snapshot of that disk.
Such a choice reduces the costs of storing such a large software in our AWS environment while making it easy and fast to create resources to launch new AlphaFold2 jobs. Moreover, it eases up the scalability of the process, so that larger prediction jobs can be launched on EC2 instances with more resources.
Once installed, AlphaFold2 is quite easy to run, requiring very few parameters to be set in the command line:
nohup python3 /data/alphafold/docker/run_docker.py --db_preset=full_dbs --model_preset=monomer_ptm --fasta_paths=/data/input/protein_sequence.fasta --max_template_date=2020-05-14
In this example, we used the provided Python script to spin-up a Docker container running AlphaFold2, provided with a protein sequence input stored in the /data/input/protein_sequence.fasta file. The parameter --model_preset can be set to multimer in order to switch the model to protein complexes prediction. After a successful run, AlphaFold2 outputs several files as well as confidence metrics. The BioStrand pipeline takes care of putting all of these within an appropriate report so that the user does not worry about the technicalities of post-processing these files.
Visualizing the output of AlphaFold2
AlphaFold2 output several models at the same time, depending on the input parameters (5 for standard run, 25 for complexes prediction). In the case of a standard run, you should find in the output repository the following files:
<target_name>/features.pklranked_{0,1,2,3,4}.pdbranking_debug.jsonrelaxed_model_{1,2,3,4,5}_pred_0.pdbresult_model_{1,2,3,4,5}_pred_0.pkltimings.jsonunrelaxed_model_{1,2,3,4,5}_pred_0.pdbmsas/bfd_uniclust_hits.a3mmgnify_hits.stopdb70_hits.hhruniref90_hits.st
Models are ranked internally by AlphaFold2, #0 being the one with the largest confidence metric. Those metrics are stored in the ranking_debug.json file, along with the ranking. Multiple sequence alignments are stored in the msas/ repository, the timings are stored in the timings.json file. You can find below an example (Fig. 1) of the values stored for each process after running AlphaFold on a random sequence (in this case, taken from an immunoglobulin domain):
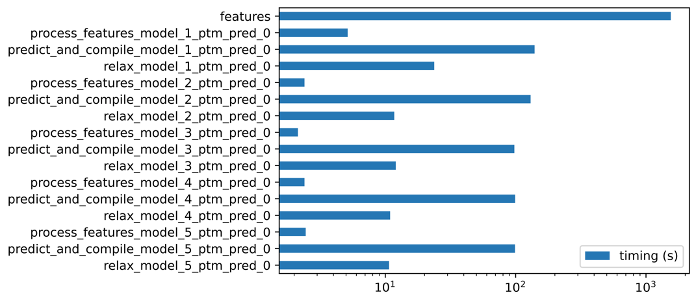 Fig. 1 - Timing of the different steps during a single AlphaFold run using an input sequence from an immunoglobulin domain.
Fig. 1 - Timing of the different steps during a single AlphaFold run using an input sequence from an immunoglobulin domain.
We can see that the step taking the largest amount of time is feature extraction (MSA), taking approximately 70% of the runtime.
The models are stored in the *.pdb files. Let’s open the 5 ranked_{*}.pdb simultaneously with a molecular viewer package, such as YASARA View. The models contain, for each residue, a local prediction confidence score (predicted LDDT score) located where the B-Factor is usually stored in models obtained from empirical methods.
After structural alignment, and coloring the models according to the predicted LDDT score per residue, we can compare them (Fig. 2):
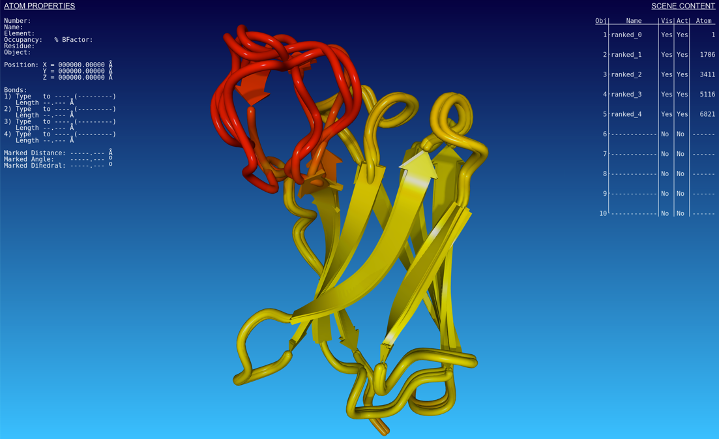
Fig. 2 - Structural alignment of predicted structures by AlphaFold2. The colour scheme corresponds to the predicted LDDT score, yellow being the theoretical maximum value.
We can see that the predictions are consistent along with the structure, except in a specific region (in red in the 3D model). In that region, the score drops to 50%, whereas it is above 90% in all other regions. Regions with predicted LDDT scores above 90% are predicted with high confidence, whereas regions with 50 % are predicted with low confidence. It is no surprise then to see different conformation predictions around that region, which must be interpreted with great care.
AlphaFold2 also outputs non-local metrics to assess model quality, which are highly relevant in order to interpret if the predicted conformation between domains in a protein complex is trustworthy or not. These are stored in the output pickle files (*.pkl), and can be read using Python code through the Pickle module of the Python Standard Library.
Conclusions
Since AlphaFold2’s reveal, many things have changed in the field of protein structure prediction. More tools and more databases are popping up left and right, making the dream of in silico drug discovery closer than ever before. While there remain improvements to be made, AlphaFold2 still provides a fast way to model proteins with capabilities beyond that of homology modeling, drastically accelerating the drug discovery process.
In the following blog post, we will discuss a bit more about the post-processing of AlphaFold2’s output and how to evaluate the predicted structure using local and non-local metrics.


