
Case study: finding robust domains in the variable region of immunoglobulins.
Searching for similarity in biological databases is easy to grasp but hard to master. DNA, RNA and protein sequence databases are often large, complex and multi-dimensional.
Conceptually simple approaches such as dynamic programming perform poorly when the alignment of multiple sequences is desired, and heuristic algorithms cut corners to gain speed.
A new method, based on advances in computer science, may combine the best of both worlds and provide great performance without sacrificing accuracy.
Searching for similarity in biological sequences is challenging
Finding patterns in biological data is one of the most important parts of many data analysis workflows in life sciences, like omics analysis. To distinguish similarity from variance is to find meaning. Whether scientists are building evolutionary trees, identifying conserved domains in proteins of interest, or studying structure-function relationships, from DNA to RNA to amino acids, they all rely on a handful of methods for finding similarity and dissimilarity in biological sequences.
Searching and aligning sequences are in its essence a problem of matching letters on a grid and assigning regions of high similarity versus regions of high variation. But nature has done a great deal to make this a challenging task.
First, there is the sheer scope of the data: the human genome contains three billion base pairs, and rarely are sequence similarity searches limited to a simple one-on-one query. Aligning genomic sequences of large patient databases means that queries become n-on-n. The simple task of matching letters on a grid of this size is computationally intensive and clever optimization is necessary, but also dangerous: cutting corners can lead to the obfuscation of meaningful data.
Apart from its size, there is another reason why biological sequence data is notoriously difficult to work with when performing alignment searches. Biological data is not static. Whenever DNA is replicated, mistakes are made. Whenever a gene is transcribed or a transcript is translated, the possibility for error arises a well.
This propensity for error is at the very heart of biology, as it is believed to be the molecular driving force behind the ability of living organisms to adapt to their environment. This elegant system of iterative adaptation however, makes biological data even more complex. Random mutations and other irregularities in biological data (SNVs, CNVs, inversions, etc.) make it difficult to differentiate between “natural noise” and meaningful differences.
All of these properties make biological datasets challenging on a conceptual and mathematical level. Even the simplest case of finding a DNA pattern in a biological database is, in a mathematical sense, not a well-posed problem. This means that possibly, no single static solution exists.
Sequence alignment: dynamic programming is slow but reliable
Many solutions to the sequence similarity-searching problem have been found. In essence, they all try to do one thing: given a set of query sequences (of any nature), find the way in which the largest number of similar or identical units (typically amino acids or bases) align with each other.
Dynamic programming is the earliest developed method for aligning sequences and remains a gold standard in terms of quality. Computationally, however, dynamic programming is sub-optimal, only being the recommended method of choice when alignments involve two, three, or four sequences. These methods are, in other words, not scalable in the least. Commonly used dynamic programming algorithms for sequence alignment are the Needleman-Wunsch algorithm and the Smith-Waterman Algorithm, developed in the 1970s and '80s.
A standard dynamic programming approach will first construct alignment spaces of all pairs of input sequences, creating a collection of one-on-one alignments that are merged together into an n-level alignment grid, where n is the number of query sequences. Although laborious, dynamic programming has the advantage of always leading to an optimal solution.
In contrast to heuristics, discussed below, dynamic programming methods do not “cut corners”, causing them to be the method of choice when a low number of sequences need to be aligned. Another advantage of dynamic programming is that it can be easily applied from open-source Python tool collections such as BioPython (https://biopython.org/), which contains the Bio.pairwise2 module for simple pairwise sequence alignment, and others for more complex alignments.
Sequence alignment: heuristics are fast but cut corners
Heuristics are defined as practical approaches to solving a data problem that do not guarantee an optimal outcome. In other words: the alignment produced by a heuristic algorithm may not be the one representing the most sequence similarity. While this sounds like a serious caveat – after all, who wants a sub-optimal solution? – their practical nature makes heuristic algorithms much less computationally intensive when compared to dynamic programming methods.
In fact, when solving complex multiple alignment problems, heuristics offer the only workable solution, because a classical dynamic programming approach to the same problem would take days or weeks of computation time.
The first popular heuristic method for sequence alignment was FASTA, developed in 1985, and it was soon followed by BLAST in the 1990s. The prime achievement of these heuristics is the use of word methods or k-tuples. “Words”, in these methods, are short sequences taken from the sequence query and matched to a database of other sequences. By performing an initial alignment with the use of these words, sequences can be offset, create a relative position alignment that greatly speeds up the rest of the sequence alignment method.
Note that the FASTA method should not to be confused with the FASTA file format, which is the default input format for FASTA alignment software, but has also become the industry standard in bioinformatics for DNA, RNA, and protein sequences throughout the years.
Many heuristics for sequence alignment are progressive methods, meaning that they build up an alignment grid by first aligning the two most similar sequences, and iteratively add less and less similar sequences to the grid until all sequences are incorporated. One pitfall to this method is that the initial choice of the “most related sequences” carries a lot of weight. If the initial estimate of which sequences are most related is incorrect, the accuracy of the final alignment suffers. Common progressive heuristics methods are Clustal and T-Coffee.
A new gold standard: the need for optimization and indexing
Neither of the two categories discussed above, dynamic programming and heuristics-based approaches, is perfect. One lacks the computational efficiency of a truly scalable tool, while the other may miss vital information. The need for a tool that combines the strengths of dynamic programming and heuristic methods, while avoiding their pitfalls, is high because databases are becoming increasingly complex and data analysis is becoming a bottleneck in many pipelines.
One way to tackle this problem is by using techniques inspired by modern computer science, such as optimization and indexing. Optimization algorithms such as hidden Markov models are especially good at aligning remotely related sequences, but still regularly fall short of more traditional methods such as dynamic programming and heuristic approaches.
Indexing, on the other hand, adopts a Google-like approach using algorithms from natural language programming to discover short informative patterns in biological sequence data, which can then be abstracted and indexed for fast retrieval on all molecular layers. Using this method, no pre-selected search window has to be specified and thus, bias is avoided. Below, a short case study is laid out, describing the search for robust domains in the variable region of immunoglobulins using HYFTsTM patterns, which allow for ultra-fast ultra-precise sequence alignment.
Case study: finding robust domains in the variable region of immunoglobulins
Immunoglobulins or antibodies are versatile, clinically relevant proteins with a wide range of applications in disease diagnosis and therapy. Complex diseases, including many types of cancer, are increasingly treated with monoclonal antibody therapies.
Key in developing these therapies is the characterization of sequence similarity in immunoglobulin variable regions. While this challenge can be approached using classical dynamic programming or heuristics, the performance of the first is poor and the latter may lead to missing out on binding sites because of the limited search window. Using indexing methods with HYFTS patterns searches the complete sequence with optimal speed.
Immunoglobulin protein sequences from PDB are decomposed into HYFTS patterns, which form fast and searchable abstractions of the sequences. Next, all sequences are aligned based on their HYFTS patterns, the outcome of which is shown below (Figure 1). The algorithm returns 900 non-equivalent sequences, which are aligned in the constant region of the immunoglobulin (red), and show more variation in the variable region (blue).
However, the variable region is not completely random, and a quick search already reveals many conserved domains in what is classically thought of as very variable domains. This search, which took less than one second and did not need any preprocessing or parameter tweaking, shows that index-based methods of sequencing alignment hold a great promise for bioinformatics, and may become the industry standard in the coming years.
For a video demonstration of this case study, see prof. Dirk Valkenborgh’s (UHasselt) talk at GSK meets universities (https://info.biostrand.be/en/gskmeetsuniversities).
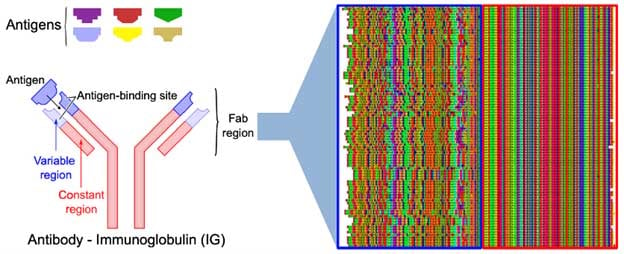
Figure 1: Alignment of immunoglobulins based on HYFTS patterns.
Conclusion
While many solutions already exist for the sequence alignment problem, the most commonly used dynamic programming and heuristic approaches still suffer from pitfalls inherent in their design. New methods emerging from computer science, relying on optimization and indexing, will likely provide a leap forward in the performance and accuracy of sequence alignment methods.
Image source: AdobeStock © Siarhei 335010335


